The Lautens group is focused on the investigation and development of novel transition-metal-mediated
organic transformations. Some projects include catalyst-controlled asymmetric transformations while
others focus on controlled tandem or domino processes.
Of particular interest are reactions which can efficiently construct frameworks of pharmaceutical compounds
or fragments of biologically-active natural products.
C-H Activation
Palladium-Catalyzed Domino Reactions with C-H Bonds: Synthesis of Substituted Carbocycles and Heterocycles using Strained Alkene Shuttles
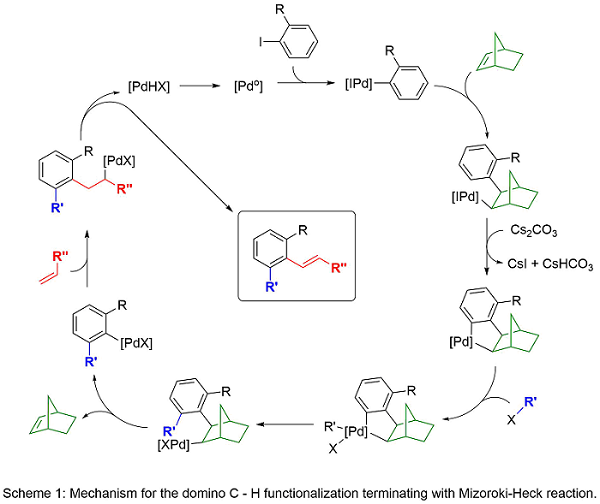
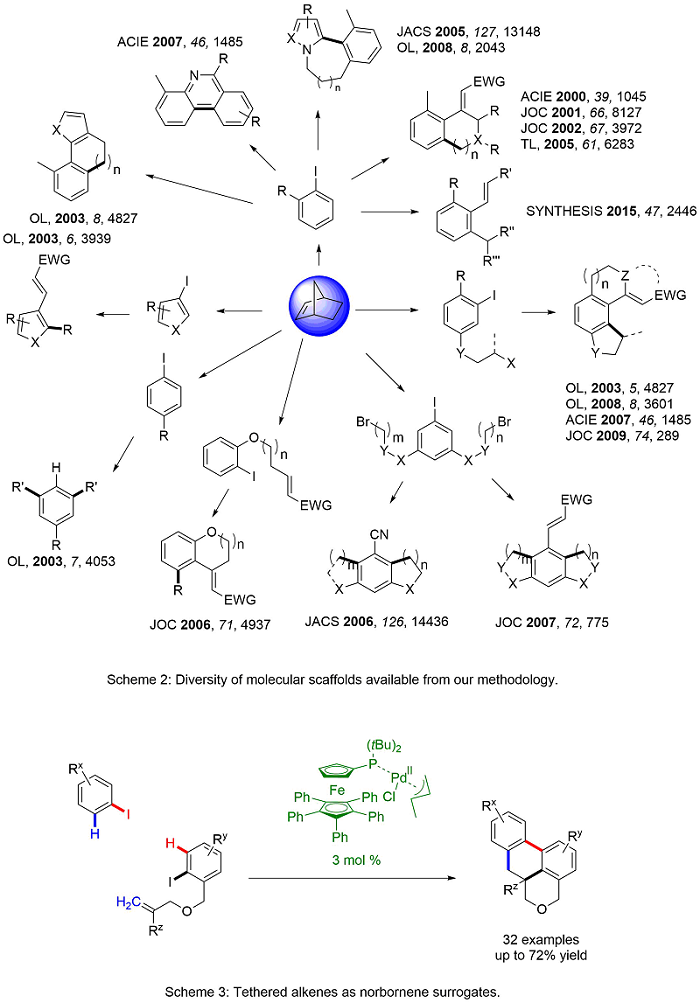
Domino reactions are efficient and cost-effective as they allow for more than one transformation in a single synthetic sequence. In 2000, we reported a palladium-catalyzed, norbornene-mediated domino reaction based on the work of Catellani1 to allow a novel synthesis of functionalized carbocycles.2 Norbornene is necessary for the reaction as it allows for the assembly of the product scaffold without being incorporated into the final product (Scheme 1). Over the years, we have modified this domino approach to include variations in the ortho C-H functionalization and the terminal functionalization steps (Scheme 2). The result is a highly general and versatile approach to functionalized carbocycles, heterocycles, and fused compounds. Recent advances has allowed the substitution of tethered alkenes in place of norbornene (Scheme 3). Current work in the area explores the synthetic diversity of the ortho-functionalization steps and the application of the methodology to new synthetic targets.
Seminal Publications
Domino C-H Activation - Heck Reaction
Domino C-H Activation - C-H Functionalisation
Domino C-H Activation - Reduction
Domino C-H Activation - With Secondary Alkyl Halides
Domino C-H Activation - Cyanation
Domino C-H Activation - Buchwald-Hartwig Coupling
Domino C-H Activation - Formation of Fused Aromatics
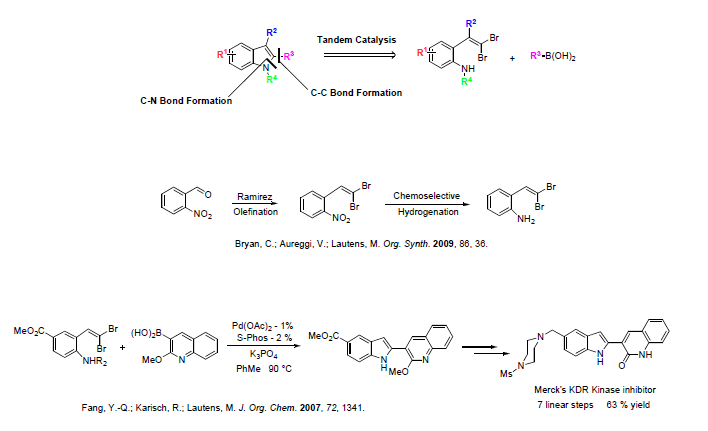
Heterocycles via Tandem Catalysis
We had envisioned the synthesis of annulated heterocycles via a tandem catalytic process in which a carbon-heteroatom bond and a carbon-carbon bond could be formed in a single operation. Indoles are a particularly attractive target of this methodology, since molecules containing this moiety are of great utility in a variety of areas. We have developed a cheap, scalable route to the necessary starting materials, where an ortho-nitro aldehyde is converted to the dibromide olefin followed by selective hydrogenation to the arylamine over a vanadium-doped platinum catalyst.1
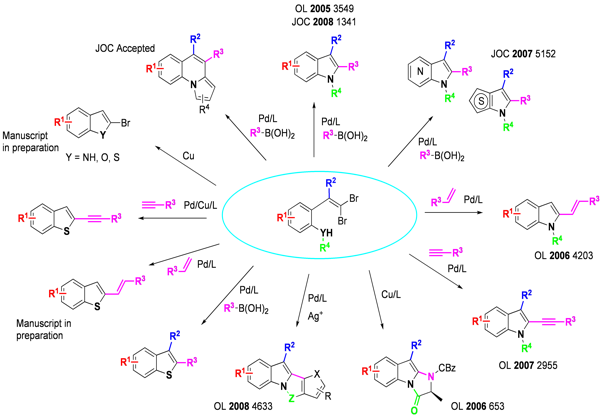
The tandem process displays high versatility: Suzuki,2,3 Heck,4 and Sonogashira5 couplings, as well as direct arylation6 can be used orthogonally to carbon-nitrogen bond formation under palladium-catalyzed conditions to create a diverse set of 2-substituted indoles, and a wide range of functionality is tolerated at all positions of the benzenoid ring. Azaindoles (including 4-, 5-, and 6- isomers) and thienopyrroles, difficult targets by traditional methodologies, can also be obtained.7 Copper catalysis is effective, as demonstrated by the synthesis of a biologically important series of imidazoindolones by tandem intramolecular amidation.8 We have demonstrated the applicability of this chemistry in the synthesis of Merck抯 KDR Kinase inhibitor.9
More recent work has focused on replacement of the arylamine moiety with other heteroatoms as well as carbon-based coupling partners,10,11 as well as the facile synthesis of 2-brominated heterocycles in the absence of a coupling partner.12
References
Rhodium-Catalyzed Enantioselective Desymmetrization
Part of our research program focuses on the development of catalytic enantioselective addition reactions to activated alkenes and alkynes. More specifically, we use rhodium catalysts to perform highly selective transformations creating new C-C bond that are not easily furnished by traditional methods. We also apply the concept of enantioselective desymmetrization for the advantage it possesses over kinetic resolution reactions in that the totality of the substrate can be converted into the desired chiral products.
A wide range of functionalized chiral alcohol building blocks can be prepared from simple starting materials bearing symmetrical allylic leaving groups. Recently, we have successfully extended the reaction to less activated linear alkenes bearing leaving groups. Prior to our work, the substitution patterns of the cyclopentenols were not accessible using existing methods. Addressing selectivity is a key element of this methodology.
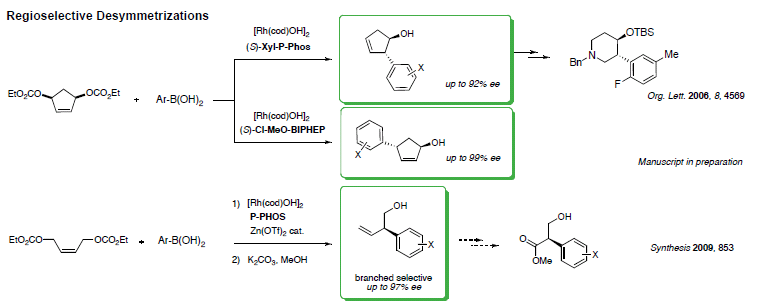
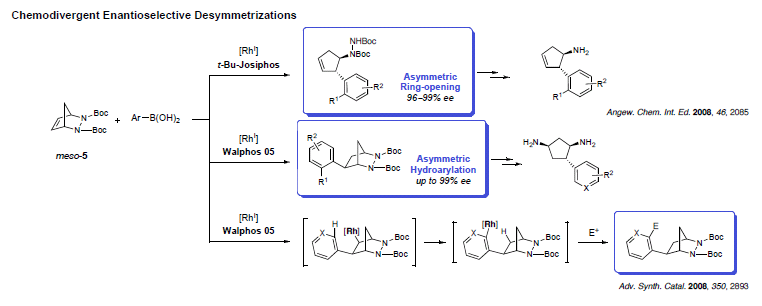
While many reactions exist for the desymmetrization of oxygenated compounds, nitrogen-containing molecules have received much less attention. Adapting our methodology for the ring-opening of diazabicycles with organometallic nucleophiles, cyclopentyl hydrazines could be prepared in high enantioselectivity. The reaction was not only successful, but also led to the unanticipated discovery of a new mechanism yielding hydroarylated products. Investigating this process in details showed that modification of the chiral ligand could favor an uncommon 1,4-Rh migration onto the aromatic ring and creating potential for further functionalization.
The strategy could be extended to a novel acylative displacement reaction. We discovered that acyl anions can be generated with a rhodium catalyst from simple boronic acids in the presence of CO. This finding makes obsolete the need for sensitive organometallic species to obtain densely functionalized carbonylated products. The unusually mild reaction condititons for this reaction (r.t., 1 atm CO) circumvents the typical need for specialized high-pressure equipment.
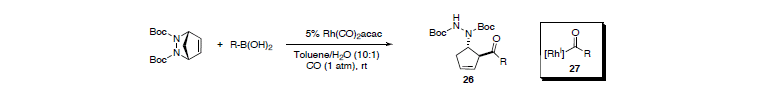
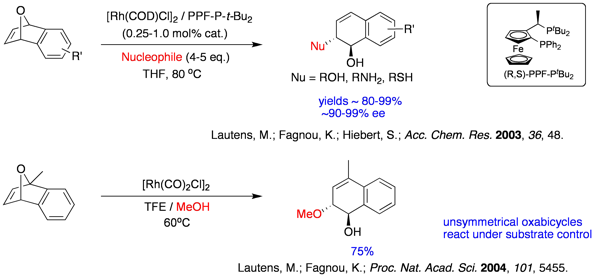
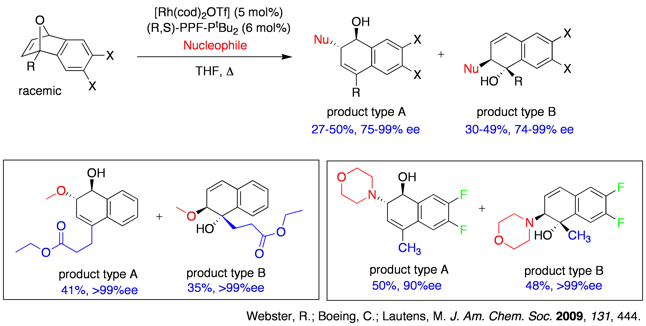
We have recently demonstrated in a rare case of utilizing water as a nucleophile in asymmetric ring opening reactions. In addition of showcasing the moisture tolerance of this ARO reaction, we also noticed a switch of diol products towards tetralones simply by increasing the catalyst loadings, which we attribute to an enantioselective isomerization reaction subsequent to the standard ring opening reaction.
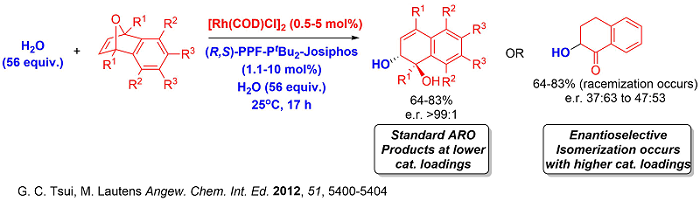
Moreover, the recent spike in interest in the synthetic community in accessing fluorinated molecules due to the prevalence of fluorine in pharmaceutically relevant compounds inspired us to design a nucleophilic fluorination ARO strategy to access fluorinated hydronaphthalenes with excellent enantioselectivities.
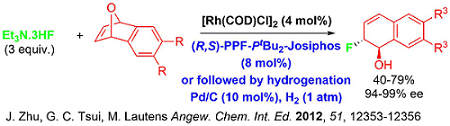
While the asymmetric ring opening reaction of oxabicyclic alkenes has been a key strategic direction of our research group for the last decade, recent investigations involved the development of optimal catalytic systems that tolerate soft carbon based nucleophiles. In 2014, our group demonstrated an instance of using cationic rhodium Rh(COD)2OTf as an effective catalyst to invoke ring opening reactions of oxabicyclic and azabicyclic alkenes with silyl-enol ethers and silyl-ketene acetals as nucleophiles. An interesting aspect of this chemistry is the observation of a silyl group transfer onto the hydronaphthalene products.
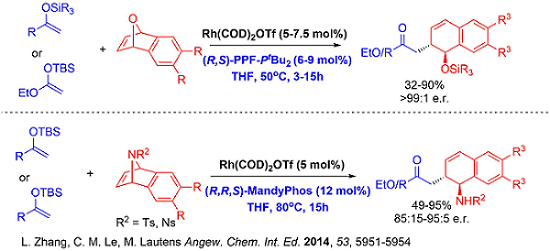
Since soft carbon nucleophiles has always been challenging using our previously developed systems, new catalytic systems in this respect would provide an efficient avenue to construct carbon-carbon bonds enantioselectively. Very recently, our group developed a fourth generation [Rh(COD)OH]2/(R,S)-PPF-PtBu2 based catalytic system that tolerates challenging carbon based nucleophiles.
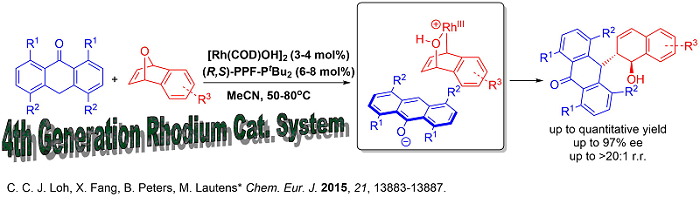
This new catalytic system paved the way for interesting benzylic functionalization involving ring opening which is hitherto unprecedented. Moreover, all other generations of catalysts for analogous oxabicycle ring opening we previously developed such as [Rh(COD)Cl]2 (1st Generation), Rh[I] (2nd Generation) and cationic Rh(COD)2OTf (3rd Generation) were unsuccessful for carbon-carbon bond formation on the benzylic position of anthrones. The protocol also showcases the first transition metal catalyzed asymmetric benzylic functionalization of anthrones attaining excellent enantioselectivities.
A unique utility of this fourth generation system was also demonstrated in a very recent publication where distal functionalization of biologically interesting cyano-coumarins was successfully executed. This is the first example in the literature where vinylogous nucleophiles could undergo ring opening reaction on any strained cyclic system via γ-reactivity.
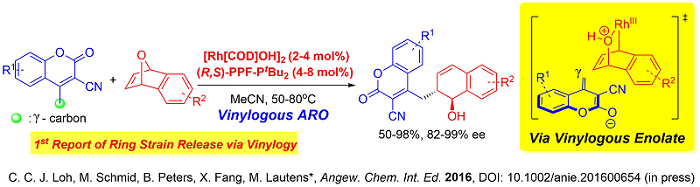
Moreover the substrate scope of this vinylogous ARO reaction is wide, tolerating even sterically hindered γ-branched cyanocoumarins. Numerous further functionalizations were demonstrated where molecular structures distinct from the parent coumarins can be accessed, effectively providing opportunities for future pharmaceutical applications. The protocol was also amenable on gram scale, further augmenting the up-scaling utility of the 4th generation catalyst system.
Total Synthesis
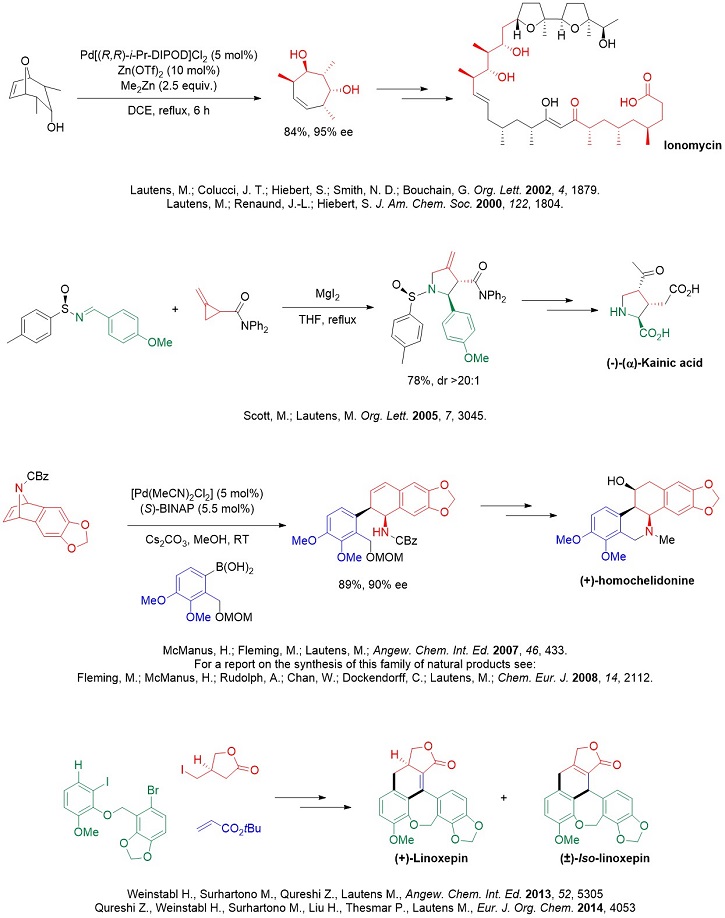
Completed syntheses:
Carbohalogenation
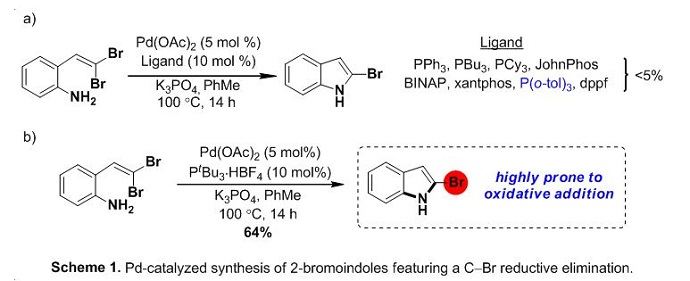
Our group envisioned that gem-dibromo olefin containing aniline derivatives could be transformed to the corresponding 2-bromoindoles via a catalytic process featuring an intramolecular Buchwald-Hartwig C−N bond formation.1 The challenge lies in the propensity of the Pd catalyst to undergo oxidative addition to the more reactive 2-bromoindole product. Therefore, a catalyst system is required which can reversibly interact with the product such that catalyst turnover is possible. In line with the observations of Hartwig and co-workers in stoichiometric mechanistic studies, the bulky monodentate trialkyl phosphine PtBu3 was shown to be essential in promoting this catalytic mode of reactivity (Scheme 1).
With this proof of concept, our group hypothesized that this mode of reactivity could exploited in a domino Heck-type carbopalladation/Csp3−halogen reductive elimination sequence.2 This approach would represent a novel complement to other methods where several classes of nucleophiles [RB(OH)2, ArSnR3, (hetero)aryl, CN-, CO/Nu-, etc.] have been used as terminating reagents for this well-established catalytic scheme (Scheme 2).
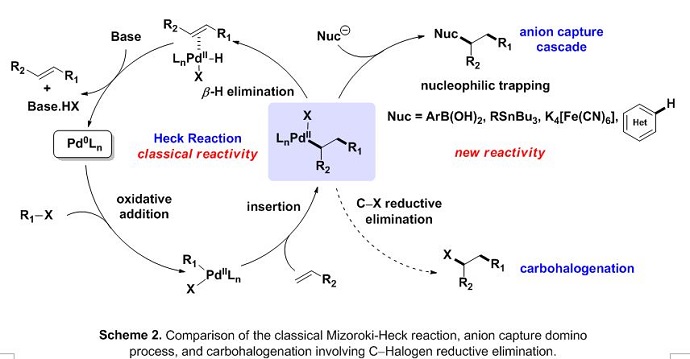
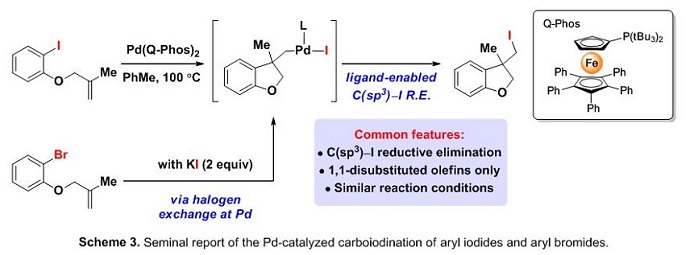
Alkene tethered aryl iodides were found to effectively undergo an intramolecular carboiodination process using PtBu3, however, Hartwig’s QPhos ligand provided an optimal reactivity profile.3 The terminating C−I reductive elimination step strictly relies on the significant amount of steric bulk possessed in this tertiary phosphine ligand. Furthermore, an anion capture derivative of this process was found to be functional under similar reaction conditions using the corresponding alkene tether aryl bromides and stoichiometric potassium iodide (Scheme 3).4
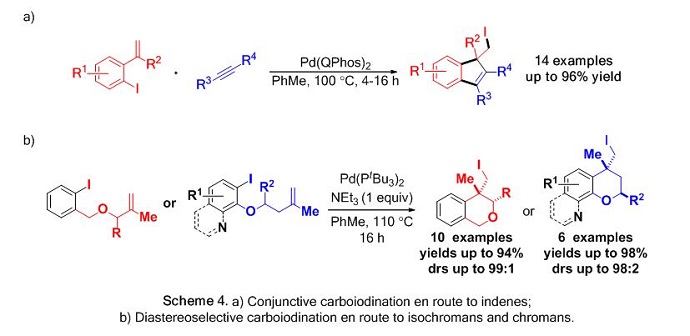
Subsequent reports highlighted the use of alkynes in a conjunctive carboiodination process en route to indenes (Scheme 4a)5 and a diastereoselective variant affording both isochromans and chromans under a single set of reaction conditions (Scheme 4b).6
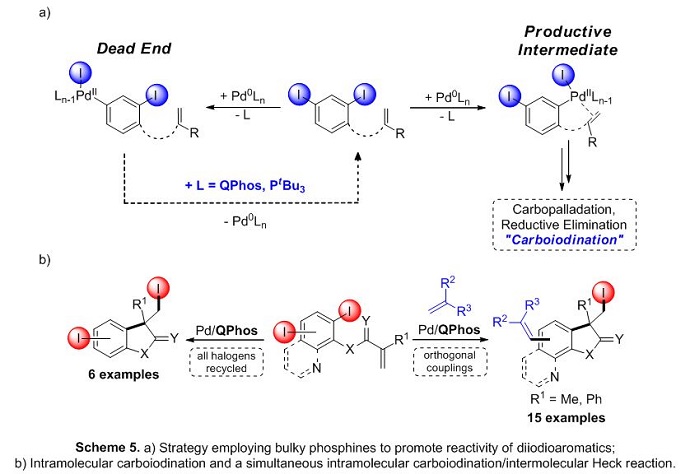
In 2013, our group was able to showcase the ability of the Pd/QPhos systems to facilitate carbon−halogen reductive elimination in a report which used diiodinated aromatic starting materials to undergo the desired intramolecular Pd-catalyzed carboiodination process. Therein, loss of catalyst activity from the formation of catalytic dead-ends arising from oxidative addition to distal C−I bonds was not observed.7 It was also shown that this specific metal/ligand combination could selectivity catalyze the simultaneous intramolecular carboiodination/intermolecular Heck reaction (Scheme 5).
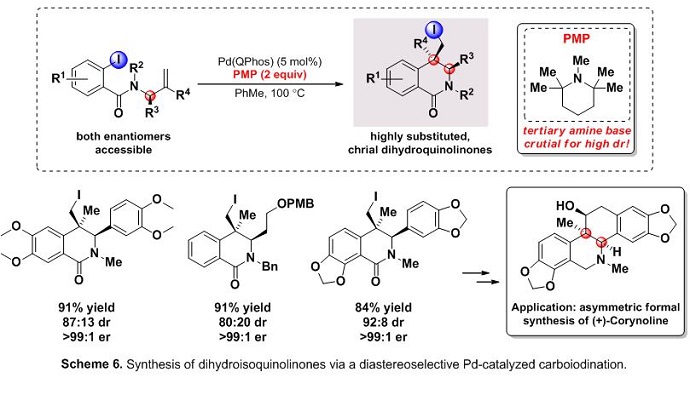
In an effort to synthesize enantioenriched dihydroisoquinolinones from chrial N-allyl carboxamides via a diastereoselective carbopalladation, our group came across an interesting effect of added amine.8 In this study it was found that dr’s could be significantly improved when the reaction was run in the presence of tertiary amine bases (i.e. PMP). This approach afforded a diverse range of enantioenriched scaffolds to be synthesized while retaining the valuable iodide functional group handle. This method was used the asymmetric formal synthesis of the alkaloid natural (+)-corynoline (Scheme 6).
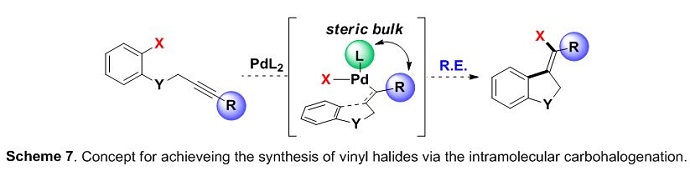
During our study concerning the use of alkynes,5 we were unsuccessful in achieving the synthesis of vinyl halides via the direct intermolecular carbohalogenation of alkynes. Later in 2015 we found that a combination of both ligand and substrate sterics were crucial achieve this class of transformation in an intramolecular fashion (Scheme 7).9
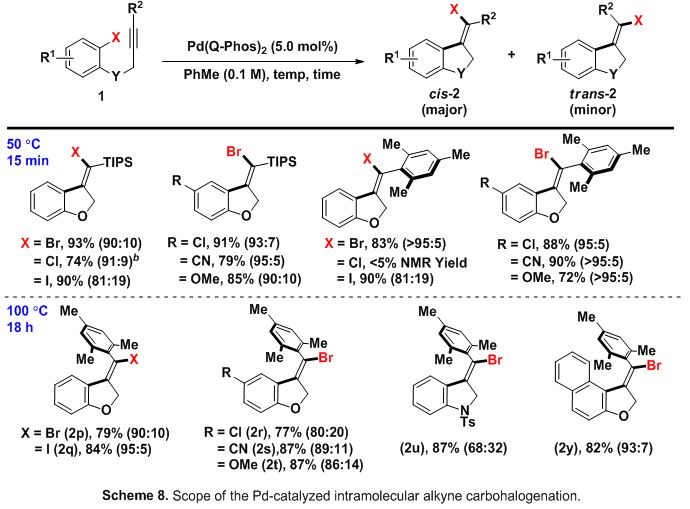
Therein, it was found that bulky groups (i.e. tBu, TIPS, TBS, mesityl, etc.) at the terminal position on the alkyne substrate, in combination with the bulky ligand QPhos were needed to effect the desired Csp2−halogen reductive elimination. When the reaction was run at 50 °C, the product resulting from a cis- carbopalladation/Csp2−halogen reductive elimination sequence was found to be major isomer, while at 100 °C (for prolonged reaction times) the major product was observed to be the apparent trans-isomer (Scheme 8). Experiments later suggested an interesting isomerization mechanism could be operative.
Domino Heck Anion Capture Cascade
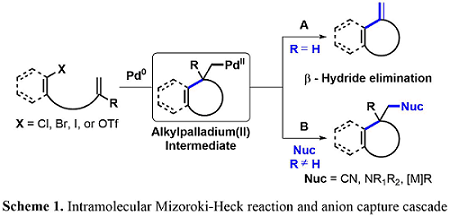
The palladium-catalyzed intramolecular Mizoroki-Heck reaction has been an interest to many groups throughout the years, since it is an effective method for installing new carbon−carbon bonds. Under standard conditions, the catalytic cycle is terminated by β-hydride elimination, which reforms an olefin moiety—a useful functional group handle in organic synthesis (Scheme 1). In the absence of a suitable β-hydrogen, the generated alkyl palladium intermediate can be terminated with varying nucleophiles resulting in a functionalized carbon quaternary center that would otherwise be difficult to install.
Following the asymmetric formal synthesis of (+)-corynoline using the carbiodination methodology of synthesizing enantioenriched dihydroisoquinolinones in a highly diastereoselective manner, the efficiency of the path to the natural product was re-examined. Within the route to (+)-corynoline, a nucleophilic displacement of the neopentyl alkyliode with KCN and 18-crown-6 ether under reflux was performed (Scheme 2). To avoid using these toxic and forcing conditions and to further increase the efficiency of the formal synthesis, the diastereoselective Pd-catalyzed arylcyanation/heteroarylcyanation was developed to furnish the desired nitrile-containing dihydroisoquinolinone.1
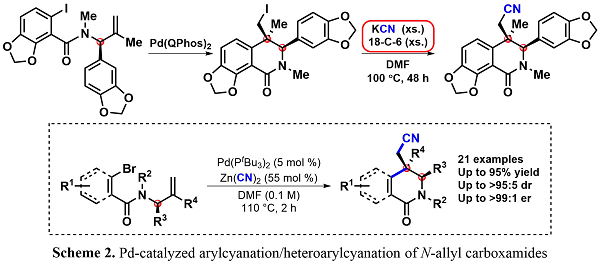
After our success in arylcyanation of N-allylcarboxamides, our group searched for a broader application of this useful reaction. Adapting our method to indoles led to the dearomative syn-1,2-arylcyanation product, thus constructing highly substituted indoline frameworks which have desirable biological activity. As seen in the reaction below, these rigid products are highly functionalized. The indoline core contains a tetrasubstituted tertiary carbon, a functionalizable acidic alpha proton, and a highly versatile nitrile group. Additionally, these scaffolds are synthesized in high yield and diastereoselectivity.2
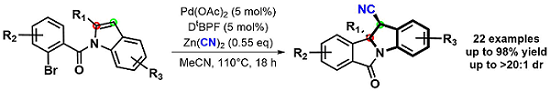
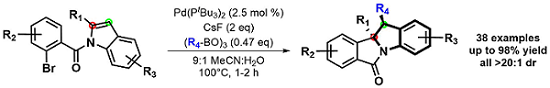
Interested in these dearomatization products, this project evolved into the study of other potential coupling partners. We developed a 1,2-diarylation of these substrates by using easily accessible boroxines. As seen below, these syn-diarylated indoline scaffolds were formed in good yields and diastereoselectivity. Moreover, the short reaction times, exceedingly broad scope and derivitizable products could allow this method to be utilized in natural product synthesis.3
Multicomponent Reactions
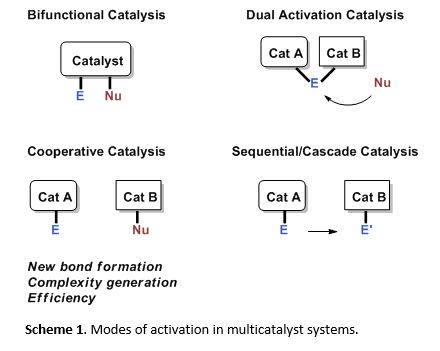
The use of multiple catalysts in a reaction vessel in organic synthesis has recently gained significant interest, as multi-catalytic systems can achieve superior reactivity and synthetic efficiency that are unmatched with single catalytic systems. Important advantages include new modes of substrate activation, new bond forming transformations, and efficient complexity generation processes that lead to waste reduction in synthesis and minimized environmental impact (Scheme 1). For example, the use of multiple metals in catalysis has provided powerful transformations such as the Wacker process and the Sonogashira cross coupling. Thus, in comparison to the traditional approach of using a single metal catalyst to perform a reaction, taking the multi-metal catalysis approach toward synthesis can be highly effective.
As ligands are integral in providing reactivity and controlling selectivity in metal-catalyzed reactions, their inclusion enables a broad range of useful transformations. The use of multiple ligands in multi-metal catalysis can facilitate optimization, improve reaction efficiency, and allow systematic ligand screening to fine-tune reactivity. Taking advantage of the dynamics and reactivity of multiple metal-ligand interactions may unlock new reactions in a variety of modes of multi-metal catalysis (Scheme 2).
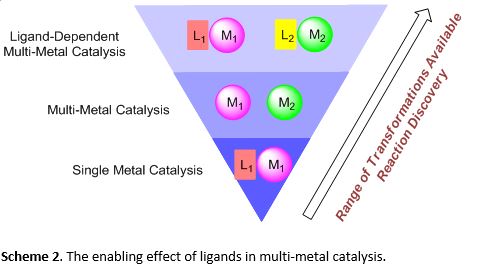
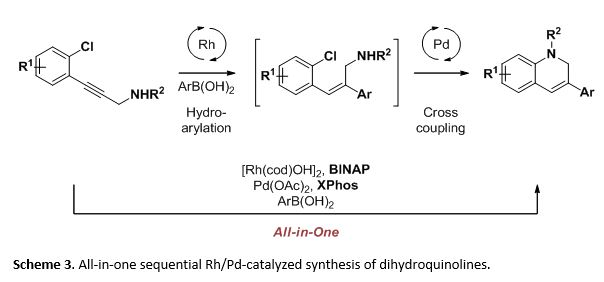
We reported the use of rhodium and palladium with a combination of two ligands, BINAP and XPhos, in the catalytic synthesis of dihydroquinolines (Scheme 3). We were interested in the Rh-catalyzed formal hydroarylation of an aryl chloride containing alkyne. In situ functionalization of the alkyne delivered an alkene for a Pd-catalyzed cross coupling. The selective formation of the desired dihydroquinoline product implied that the reaction pathway must be highly controlled. Our investigation of the mechanism led us to two observations that were important for the development of multi-metal-catalyzed reactions.
Having both catalysts and ligands in at the same time, a number of undesired transformations could occur under the reaction conditions. For example, the presence of arylboronic acids, arylhalides, and palladium could lead to an undesired Suzuki-Miyaura cross coupling (Scheme 4). Consequently, a time-resolved reaction sequence with defined catalytic cycles was clearly present in this sequential process.
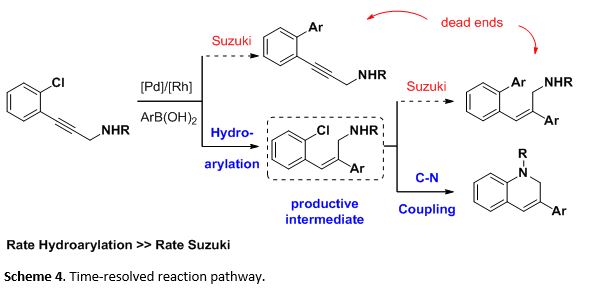
Our studies on the relative rates of Rh and Pd catalysis led to the conclusion that the fast Rh-catalyzed hydroarylation minimized the Suzuki cross coupling, funneling the reaction pathway to the productive intermediate, which then underwent the C-N coupling to afford the desired products.
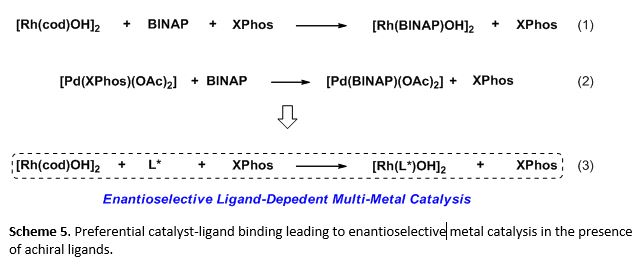
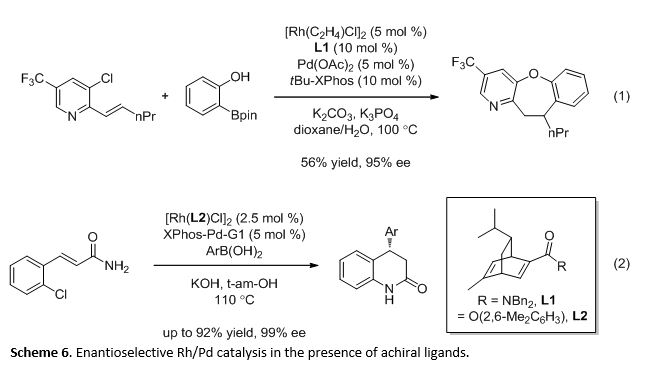
In the multi-catalytic system, Rh and BINAP were essential for catalyzing the hydroarylation and Pd and XPhos were essential for catalyzing the C-N coupling respectively. However, Pd and BINAP were ineffective at promoting the C-N coupling. As a result, we probed the binding behaviors of the catalyst-ligand mixtures (Scheme 5). We found a selective association of Rh for BINAP over XPhos (eq 1). While Pd does associate with BINAP and XPhos, there was also a preferential binding with BINAP (eq 2). However, the selective Rh-BINAP association was crucial in minimizing the formation of Pd-BINAP, an inactive catalyst-ligand species. Furthermore, the preferential association led us to the use of chiral ligands for Rh catalysis in combination with Pd and XPhos in the development of enantioselective metal catalysis in the presence of achiral ligands (eq 3).
As ligands are versatile and confer reactivity and selectivity, various compatible combinations of ligands can be used to promote different reactions using the same metal pairs. Thus, we are interested in demonstrating the ability of ligands in multi-metal catalysis to achieve a greater range of transformations that could be developed. For example, by tuning the ligands and reaction conditions, we were able to achieve Rh/Pd-catalyzed synthesis of benzoxepines (Scheme 6, eq 1) and dihydroquinolinones (eq 2) in high enantioselectivities.
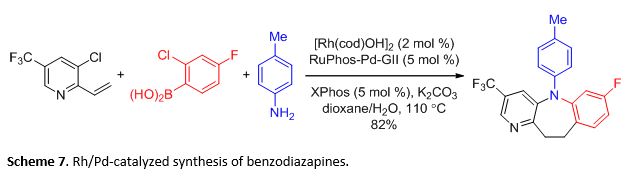
Multicomponent reactions (MCR) assemble three or more reactants in a controlled manner to afford a single product that retains the majority of atoms in the reactants. Due to the broad scope that MCRs can achieve, these reactions are highly useful in combinatorial chemistry and diversity oriented synthesis. Libraries generated from MCRs can be assayed in high throughput screening to look for desired biological properties. Thus, MCR remains a common and important tool used in drug discovery, often employed to develop new leads. As the use of multiple metal catalysts significantly enlarges the number of transformations available in reaction design, the potential for developing new MCRs based on multi-metal catalyst systems can be realized. For example, our group recently reported the use of a combination of Rh and Pd that could successfully stitch together a vinyl pyridine, o-chloroaryl boronic acid, and an amine to access benzodiazapines (Scheme 7). This (MC)2R exhibited a remarkable control in the reaction pathway. Given the number of reactive functional groups present that could lead to various undesired pathways, the time resolution conferred by Rh-catalysis was crucial in the success of this reaction.
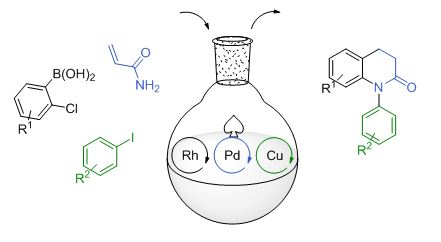
With the aim to develop (MC)2R based on the compatible, versatile, and robust Rh/Pd system, we further expanded the catalysis with the incorporation of copper. Following a Rh/Pd-catalyzed conjugate addition/C-N cross coupling, we performed a subsequent a Cu-catalyzed Goldberg amidation in a one pot manner to access N-aryl dihydroquinolinones (Scheme 8). Members of this class of products exhibit a number of important biological properties, including Aripiprazole, the second top selling drug in 2012.
Zhang, L.; Sonaglia, L.; Stacey, J.; Lautens, M. Org. Lett. 2013, 15, 2128-2131.